
Top tips for providing the right amount of detail in first-in-human common technical documents
Category | Large Molecule
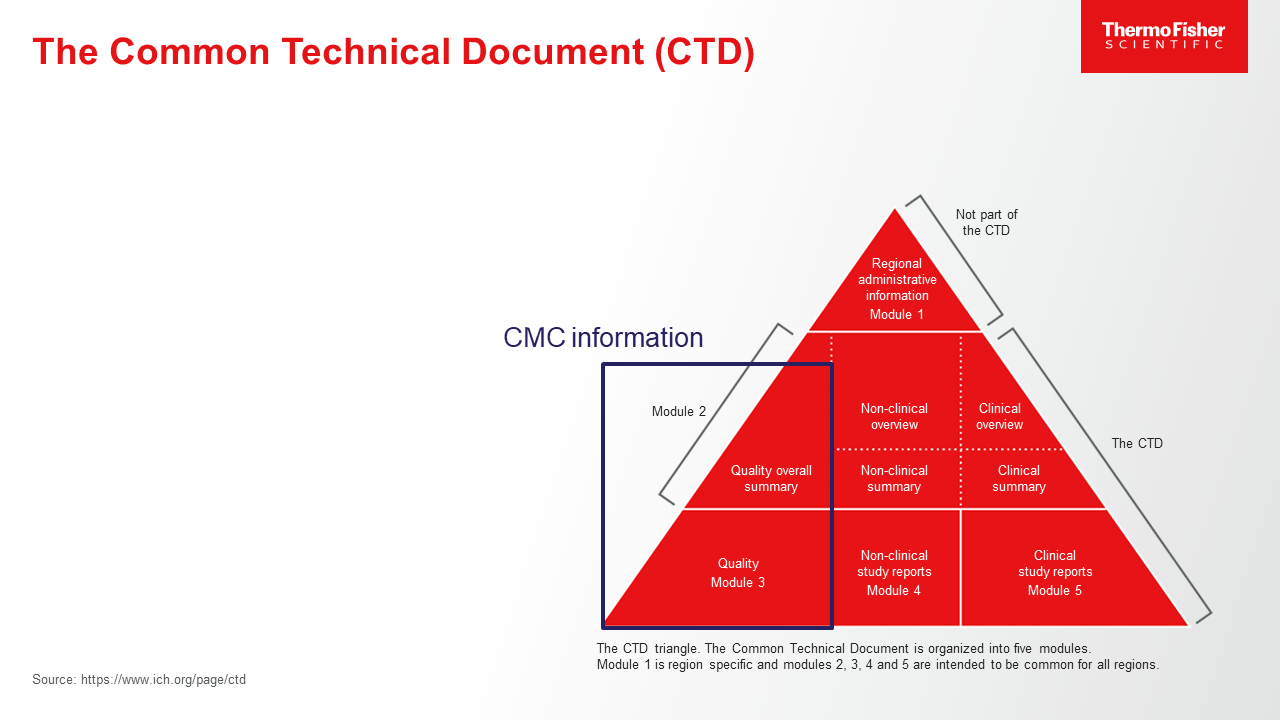
In the early-development stage, little may be known about a drug’s characteristics. What’s more, drug processes and formulations frequently evolve as more information emerges following testing and trials. As companies prepare to enter first-in-human (FIH) studies, they often face uncertainty regarding how much information will be expected in sections of the common technical document (CTD),1 such as information related to chemistry, manufacturing, and controls (CMC, CTD Module 3) for a FIH clinical trial application. Typically, only limited data are available at this phase of development, leaving sponsors wondering how much detail to include in the dossier. Here are some guidelines to follow regarding the structure and content of a FIH clinical trial application dossier.
1. Focus on human safety when discussing manufacturing processes, controls, and reference standards
FIH clinical trial application dossiers in the CTD format must spell out the details of the current manufacturing process and provide reasoning for the selection of a specific approach, process, or method, as well as for any changes from prior methods. A prospective comparison of the preclinical and clinical materials based on predetermined measures will be expected anytime a process parameter change or manufacturing adjustment could impact characteristics such as purity, potency, and other qualities closely tied to safety.
A high-level description of the process is typically sufficient at this stage of development, as long as regulators can make a basic evaluation of the process’s performance and the expected safety and quality of the drug substance and drug product. While reference standards are expected to be evolving and no validation information is required for FIH studies, the dossier should:
- Provide evidence that the manufacturing process can produce a sterile product of appropriate quality
- Highlight known molecule characteristics and relate them to safety, efficacy, and human suitability factors
- Share quantitative results when possible
- Summarize analytical data and release methods
In summary, the CTD should provide a high-level description of the process and discuss which parameters will be monitored and considered as indicators in early development, as drug substance manufacturing process and controls are still under development at the FIH stage.


2. Share characterization and description details that support a transition to human research
As critical quality attributes (CQAs) about a drug often have not yet been selected at this phase, it will be essential to address them in the dossier. The primary concern is whether the drug product is suitable in all features for transition to trials with human beings. The dossier should:
- Acknowledge current limitations in knowledge. As CQAs are identified through additional characterization and process development, the dossier can be amended and updated.
- Describe all that is known regarding drug characteristics which heavily impact quality and safety, such as purity, potency, strength, and clearance.
- Provide quantitative results from potency and purity assays to demonstrate product quality.
TIP: Include the following:
- Summary for each unit operation to assure sterility
- Summary information on formulation development, including stability, compatibility, and novel excipients
- Limits for sterility and particulates
- Limits for residual DNA, residual host cell protein (HCP), and endotoxin
3. Discuss preliminary control parameters and stability assessment plans
Ensure that the submitted stability data support at least the planned clinical study duration.Support your claims with available test results.2,3
In addition, the stability section should:
- Provide any results from accelerated degradation and stress condition testing
- Describe quantitative stability and degradation information from preclinical or non-GMP (good manufacturing practice) batches, when available
- Share details on timepoint planning for further stability studies
TIP: Cell line details are critical.
Provide thorough information about host cell line origin, expression construct derivatization, and cell banking. A working cell bank is not required at this point, but there must be sufficient material to complete the planned studies.
4. Include essential details for submission
Not matter what phase - I, II or III - key essentials for a successful clinical trial dossier submission include:
- Highlight how each element may impact human safety. Changes to manufacturing processes, identification and adjustment of critical parameters, and key attribute testing must all be discussed in the context of suitability of the product for human use.
- Build the document with a forward-thinking strategy and prepare each draft with next steps in mind.
- Show the thought process behind product development. Spell out the plan and reasoning behind changes to convince regulators of the strategy and appropriateness of decisions.
- Involve regulatory personnel early and often to gain feedback on what their concerns and recommendations will be for the drug and its production.
In conclusion, it is critical to be able to track and justify all changes made during development, particularly as the drug advances through human trials. At all stages the CTD’s primary purpose is to provide compelling, cohesive evidence to regulators that the drug is ready for the next level of human research or approval.
Further details on what to include in CMC sections of the CTD—and in what depth—can be found in our printable checklist or our webinar.
To learn more about how Thermo Fisher Scientific fosters its digital culture and how you can begin your organization’s digital transformation, click here to access our new checklist.
View references
