The information you provide to the chat will be recorded to improve your experience and to contact you. Please read our privacy notice to see how we are processing and protecting your data. Click to view our Cookie Notice.
We'd love your feedback—take a quick survey to help us improve.
How can we help you today?

Pharma insights blog
Articles by technical and scientific subject matter experts

Blog post
Beyond the Tufts study: Real-world application of CDMO–CRO integration
Learn how Tufts CSDD benchmark data translates into integrated CDMO–CRO execution by reducing handoffs, silos, and development delays.
13 minute read

Blog post
Clinical trial logistics is becoming a strategic discipline: What biopharma needs to prepare for next
Clinical development is becoming more global, complex, and dynamic—yet the systems that move materials, data, and decisions are still often treated as background operations. This shift is elevating clinical logistics from “plumbing” to a strategic discipline that directly influences speed, quality, and resilience across development programs.
13 minute read

Blog post
The real challenge of biologics scale-up isn’t capacity—it’s demand
Most biologics never reach the production volumes that justify large-scale stainless-steel capacity. The real challenge isn’t building enough—it’s knowing how much will truly be needed, and when. Here’s why flexibility in scaling is becoming one of biologics manufacturing’s most strategic advantages.
16 minute read

Blog post
How to Achieve Late-Phase Success in OSD Development
Learn how to achieve late-phase success in oral solid dose scale-up with data-driven strategies, risk mitigation, and the right CDMO partnership.
11 minute read

Blog post
CPHI Frankfurt: A leader’s perspective on how strategic integration drives progress in drug development
Integration, digital enablement, and quality are shaping the next phase of biopharma partnerships. In a presentation at CPHI Frankfurt 2025, Jennifer Cannon, President of Commercial Operations at Thermo Fisher Scientific, challenged the audience to rethink how value is created in today’s high-pressure biotech environment, as companies face mounting investor expectations, tighter funding cycles, and growing regulatory complexity.
10 minute read

Blog post
CDMO Supply Chain Excellence with Visibility & Transparency
Discover why real-time data, operational visibility, and transparency throughout the supply chain are essential for operational CDMO excellence and client satisfaction.
10 minute read

Blog post
From promise to patients: Smarter pathways to scale in cell and gene therapy
Cell and gene therapy’s future depends not only on scientific breakthroughs but on how effectively they are scaled. Progress will come from rethinking traditional models—combining flexibility, standardization, and collaboration to move innovative therapies from promise to patient impact. Read the blog to explore how new approaches are reshaping the path from discovery to delivery.
12 minute read

Blog post
Avoiding regulatory setbacks in OSD development: Why early formulation decisions matter
Early formulation choices can shape the entire trajectory of an oral solid dose program. Missteps in areas like bioavailability, stability, or regulatory documentation often surface downstream, creating costly delays. Aligning formulation strategy with scalability and compliance from the start helps ensure readiness from IND to NDA.
10 minute read

Blog post
Early-stage biotech: The wrong outsourcing strategy costs more than time
For early-stage biotech companies, outsourcing decisions can make or break program momentum. A strategically aligned development model that connects early planning with downstream execution is critical to avoiding delays, reducing risk, and building long-term value.
10 minute read

Blog post
Expert perspective: Navigating regulatory challenges in ancillary supply management
Explore expert perspective on what regulatory challenges are faced in ancillary management, and how to best minimize risk and adapt to evolving requirements.
15 minute read
Blog post
Understanding the essential role of Qualified Persons in clinical trials
Navigating complex supply chains while adhering to regulatory compliance in clinical trials can be a challenge. With differences between the UK and EU certifications, Qualified Persons (QPs) play a crucial role in ensuring the highest quality and safety standards. Read this blog post to learn how a strong QP partnership is often necessary for investigational products to reach patients on schedule.
11 minute read

Blog post
Is your API manufacturable—or just technically feasible?
Treating manufacturability as a late-stage concern can lead to costly delays, rework, or even failure. This post explores why early investment in process design is critical—and what manufacturability looks like in practice at Thermo Fisher Scientific’s API center of excellence in Cork, Ireland.
12 minute read

Blog post
Expert perspective: Advancing ultra-low temperature labels for advanced therapies and vaccines
Navigating the complexities of ultra-low temperature labeling for advanced therapies and vaccines is a challenging endeavor. The COVID-19 pandemic has highlighted the need for reliable clinical labeling solutions that can withstand ultra-low temperatures. Julia Field, Sr. Product Manager at Thermo Fisher Scientific, provides expert insights into their innovative ultra-low temperature labeling offerings, highlighting the importance of these advancements in maintaining the safety and efficiency of the clinical supply chain.
15 minute read

Blog post
Expert perspective: How microdosing in clinical trials is accelerating drug development
Learn from our experts, Jutta Wagner and Christian Rose, on how microdosing in clinical trials is accelerating drug development.
15 minute read

Blog post
Accelerate drug development by embracing an integrated approach
Pharmaceutical companies face significant challenges with rising R&D costs and extended development timelines. Embracing an integrated CDMO and CRO approach that combines drug substance, drug product, clinical manufacturing, clinical research, and clinical supply chain management into one streamlined service can simplify complexity, enhance efficiency, and reduce risks, ultimately accelerating the journey from lab to market.
12 minute read

Blog post
Discussing the Future of Biotech
Explore expert insight into what trends are shaping the biotech industry, including innovations, sustainability, AI, and growth in a dynamic landscape.
10 minute read

Blog post
Five hidden risks in early-phase OSD formulation development
Early-phase oral solid dose development presents a range of hidden risks that can impact long-term success. From API complexity to scalability and regulatory readiness, thoughtful planning can help mitigate setbacks and keep programs on track.
12 minute read
Blog post
Streamlining biological sample management: The advantages of onsite sample processing and biorepository integration
Discover how integrating sample processing and biorepository storage in one location can accelerate research, protect sample integrity, and reduce operational costs. This blog post outlines key benefits—from faster turnaround times to enhanced flexibility and access to specialized lab networks.
12 minute read
Blog post
Rethinking 'Platforms’ in Cell and Gene Therapy Development
Discover why developers are looking beyond predefined platform processes and toward flexible customization, like our Rapid Development Framework.
14 minute read

Blog post
Enabling flexibility and scientific innovation in oral solid dose development
A strategic CDMO partnership enabled a clinical-stage pharmaceutical company to navigate the complexities of early drug development, advancing formulation strategies, adapting to shifting priorities, and ensuring a scalable path to clinical trials.
12 minute read

Blog post
Rethinking biologics storage: Navigating nontraditional temperatures
Learn how the right cold chain storage provider can protect your biologics from nontraditional temperature storage requirements
9 minute read

Blog post
Decentralized clinical trials: A game changer for biotech drug developers
This blog examines the rise of decentralized clinical trials in the pharmaceutical industry and explores why they’re a game changer for biotechnology drug developers of all shapes and sizes.
10 minute read
Blog post
Expert perspective: How Thermo Fisher Scientific’s packaging and labeling innovations simplify biotech clinical trials
Find expert insight on how innovative clinical packaging and labeling solutions can streamline processes, enhance efficiency, compliance and overall success.
15 minute read
Blog post
The science of cell line development for biologics: Improving stability and yield
Cell line development is critical to biologics manufacturing, influencing efficiency, scalability, and product stability. Advances in CHO-K1 cell line engineering, are driving higher yields, improved gene stability, and faster IND submission pathways.
15 minute read

Blog post
High-potency OSD development solutions: View from Bourgoin.
Explore examples of how biotech and pharma companies leverage our strategically located European facility to solve high-potency API and OSD challenges.
14 minute read
Blog post
Advantages of localized Oral Solid Dose production in Europe
Discover how partnering with a localized OSD production facility in Europe can offer advantages, including regulatory expertise and supply chain stability.
15 minute read

How Clinical Packaging and Labeling Innovations Drive Value
Effective clinical packaging and labeling drive value by ensuring compliance, protecting product integrity, and improving trial efficiency. A strategic approach helps biotech sponsors mitigate risk, adapt to trial demands, and optimize cost, speed, and reliability.
15 minute read
Blog post
Expert perspective: Navigating the complex path of biologics manufacturing:
The journey from preclinical development to clinical manufacturing in biologics is as intricate as the therapies it aims to deliver. For biopharma companies, understanding the critical steps and considerations of this process can mean the difference between delays and success. Otto Jurrius, General Manager of Thermo Fisher Scientific’s biologics manufacturing facility in Groningen, Netherlands, provides expert insights into the challenges and innovations shaping this complex landscape.
15 minute read
Understanding large molecule drugs
This blog provides a deep dive into large molecule drugs, or biologics, exploring their key characteristics, advantages and challenges, and future in the pharmaceutical industry.
18 minute read

Blog post
The critical role of cold chain logistics: Safeguarding drug integrity from lab to patient
This blog explores the critical role of cold chain logistics in the biopharma industry and explains how a CDMO/CRO partner can help drug developers safeguard the integrity of their products.
(12 minute read)

Blog post
Tamper-evident Packaging for Clinical Supply Protection
Thermo Fisher Scientific’s patented Tamper Evident Carton provides a comprehensive answer to tamper-sealing concerns in clinical supply chains. This design eliminates the need for traditional tamper-evident labels by incorporating the carton, insert, and tamper-evident features into a single, unified package.
(12 minute read)

Blog post
Scaling allogeneic cell therapies: Overcoming manufacturing hurdles
Scaling allogeneic cell therapies requires overcoming unique manufacturing hurdles to achieve safe, high-volume production. This article explores key challenges and best practices to maintain quality and efficiency, making these transformative therapies more accessible to patients worldwide.
(15 minute read)

Blog post
Redefining acceleration of the drug development journey
In the pharmaceutical industry, getting essential treatments to patients quickly and safely demands a comprehensive approach that reduces delay, complexity and risk throughout the drug development journey. Accelerator™ Drug Development, Thermo Fisher Scientific’s 360˚ CDMO and CRO solutions, brings all those necessary services under one roof, enabling customers to achieve their goals of getting treatments to patients faster.
16 minute read

Blog post
What are Small Molecule Drugs?
Explore the ins and outs of small molecule drugs, including key development and manufacturing challenges, as well as their future potential in modern medicine.
16 minute read

Blog post
Insights from CPHI 2024: Navigating the high costs of cell and gene therapy development with flexible financial solutions
Cell and gene therapy developers face significant financial challenges that can stall critical projects. Flexible financial solutions are helping companies manage costs and advance their therapies. During CPHI 2024 Milan, Kelly Howard, Vice President of Commercial Operations for Viral Vector, mRNA, and Cell Therapy Services at Thermo Fisher Scientific, discussed flexible options available through Thermo Fisher Financial Solutions.
8 minute read
Blog post
Manufacturing autologous cell therapies: challenges and best practices
Manufacturing autologous cell therapies is a complex and resource-intensive process that involves unique challenges like supply chain complexity, scalability, and high costs. These therapies, which utilize a patient's own cells, require a highly personalized approach at every stage. However, by adopting best practices—such as optimizing supply chain management, implementing scalable manufacturing solutions, and staying aligned with regulatory requirements—manufacturers can enhance efficiency and reduce costs, making these life-saving therapies more accessible to patients in need.
(17 minute read)

Blog post
Emerging trends in cell therapy: Autologous and allogeneic perspectives
The cell therapy sector is experiencing significant growth, driven by innovations in biotechnology and increased investment in research and development. Despite the promise of these therapies, the intricate manufacturing processes for both autologous and allogeneic treatments present significant challenges. Thermo Fisher Scientific’s commitment as a manufacturing partner is to stay at the forefront of these advancements, driving innovation and ensuring the highest standards in cell therapy development and manufacturing.
10 minute read
Blog post
Understanding cell therapies: Key differences between autologous and allogeneic approaches
By harnessing living cells to repair, replace, or regenerate damaged tissues and organs, cell therapy offers personalized and potentially curative treatment for a wide range of conditions. As the field evolves, understanding the nuances between different types of cell therapies is crucial for professionals involved in the development and manufacturing of these therapies.
12 minute read

Why process development matters: Six benefits for biotech and pharma companies
This blog explores the essential role of process development in drug development and manufacturing, highlighting six key benefits that sponsors can gain by prioritizing this task.
15 minute read

Embracing green chemistry for sustainable API development and manufacturing
Early-stage API development, despite its inherent risks and uncertainties, is the critical period for embedding green principles. This proactive approach not only reduces environmental impact but also lays the foundation for scalable and cost-effective processes as the compound progresses through clinical phases.
9 minute read

Blog post
The power of partnership in small molecule discovery and development
Pharma companies can streamline their small molecule drug development by leveraging the internal synergies of a single, trusted provider for discovery resources and development and manufacturing services.
9 minute read
Blog post
Advancing viral vector development through Quality by Design approach
Explore more on NysnoBio's partnership with Thermo Fisher, focusing on a quality by design approach to viral vector development and manufacturing.
12 minute read

Blog post
Meeting biopharma challenges: An insider’s view on CDMO solutions for biologics
Maider Parikh, Ph.D., Vice President, Commercial Operations, Biologics at Thermo Fisher Scientific discusses the key biopharma challenges including meeting project timelines, navigating regulatory requirements, and ensuring supply chain reliability, and explains how advanced technologies, expertise, and strategic planning enable Thermo Fisher to address these challenges and help bring biologic products to market faster.
9 minute read
Tech transfer, part 2: The value of strategic partnerships in technology transfer
Strategic partnerships play a crucial role in successful technology transfers by helping to identify and mitigate process risks. A well-chosen strategic partner can preserve project timelines, overcome common technology transfer challenges, and lead to significant cost savings.
10 minute read
Tech transfer, part 1: Navigating uncertainty in pharmaceutical manufacturing: The critical role of technology transfer
Efficient technology transfer in pharmaceutical manufacturing helps maintain product quality, protect intellectual property, manage costs, and scale operations, thereby ensuring that companies can respond effectively to new opportunities or challenges, maintain competitive advantage, and ensure uninterrupted supply of medications to patients.
15 minute read

Five best practices for integrating drug substance and drug product development
Discover five best practices to consider when integrating drug substance and drug product development to optimize the efficiency and effectiveness of your approach.
9 minute read

CDMO quality harmonization: Ensuring consistency, reliability, and supply chain resilience
The harmonization of quality standards, procedures, and practices across sites is crucial for enhancing the resilience and efficiency of the entire supply chain and is a key driver in accelerating market entry for safe, effective therapies.
14 minute read

What’s hot in freeze drying? Your lyophilization questions answered
Lyophilization is a critical process in the sterile fill-finish phase of pharmaceutical manufacturing, particularly for products that require high levels of stability and a longer shelf life. The ability to transform drug products into a dry powder without compromising their structural integrity is particularly crucial for preserving the stability and efficacy of biologic products, such as vaccines, antibodies, and other protein-based therapies. Over the years, advancements in technology and process optimization have made lyophilization more efficient and reliable for a wide range of pharmaceutical applications.
15 minute read

Revolutionizing drug development: AI-driven solutions for solubility and bioavailability challenges
The integration of AI/ML technologies in drug development, particularly in addressing solubility and bioavailability challenges, marks a significant paradigm shift in the pharmaceutical industry. These advanced computational methods are transforming the traditional resource-intensive trial-and-error processes into more efficient, accurate, and cost-effective strategies.
12 minute read

Mastering complex small molecule APIs and formulations
Complex small molecule APIs are characterized by their intricate structures, higher molecular weights, and sophisticated delivery requirements, which pose unique challenges in their formulation and manufacturing processes.
15 minute read

Predictive modeling for solubility and bioavailability enhancement
Explore the challenges faced by poor solubility and low bioavailability in pharmaceutical formulation and the potential of predictive modeling to overcome these challenges.
15 minute read

The quality lever: Shaping success in CDMO partnerships
Compromising on quality can lead to detrimental impacts on both speed and cost, ultimately affecting the successful development and marketing of new therapies.
10 minute read

Blog post
A CDMO Partner for every Gene Therapy Manufacturing Stage
Take a closer look at the experiences of NysnoBio and bluebird bio for insight into what companies need in a CDMO partner for every stage of viral vector manufacturing and development.
20 minute read

Blog post
CDMO 2.0: Three pharma industry trends for 2024 and beyond
Discover three major trends expected in the pharma industry, including turning to flexible manufacturing, embracing digital enablement, and the need for CDMOs deliver transformational value.
15 minute read

Blog post
Benefits of an integrated approach to viral vector manufacturing
Learn why biopharma companies choose to partner with integrated CROs and CDMOs with experience developing and manufacturing viral vectors over doing the work in-house.
15 minute read
Blog post
Understanding the viral vector product journey
Learn why biopharma companies choose to partner with CDMOs to leverage their innovative, integrated, and ready-to-use solutions for viral vector development and manufacturing for gene therapies.
20 minute read

Blog post
Exploring four patient-centric trends shaping today’s biopharma landscape
The biopharma industry is adopting a patient-centric approach to drug research, development, and manufacturing. Explore four trends shaping today’s landscape.
15 minute read

Blog post
Sterile injectable therapies: Changing delivery formats revolutionizes lifecycle management
The sterile injectable drug market is evolving at a rapid pace. This segment's significance cannot be understated. A closer examination reveals the intricacies of this evolving landscape, specifically in the realm of delivery formats.
8 minute read

Transforming clinical trial supply chain optimization through digitization
Innovative technologies such as artificial intelligence, automation, and real-time data analytics stand to revolutionize clinical trial supply planning from an efficiency, accuracy, and resiliency standpoint.
15 minute read

Blog post
Key Insights from CPHI Barcelona 2023
As the largest global event for pharmaceutical supply chain companies, CPHI is a microcosm of the pharmaceutical manufacturing industry. At this year’s event, the rapidly shifting pharma landscape contributed to an undercurrent of urgency. Following are some of the key takeaways.
10 minute read

Blog post
Inside pharmaceutical formulation development
Pharmaceutical formulation is a key aspect of drug development and helps to ensure safe, effective, and patient-friendly medications for people worldwide.
15 minute read

Blog post
Unlocking efficiency: Pros and cons of outsourcing your biorepository
When it comes to biorepositories, should biopharma companies insource or outsource their storage needs? This blog breaks down the pros and cons.
8 minute read

Blog post
Building a biorepository: Weighing the benefits and drawbacks
Biorepositories play a crucial role in collecting, preserving, and utilizing biological materials, but does it make sense to build or buy one?
8 minute read
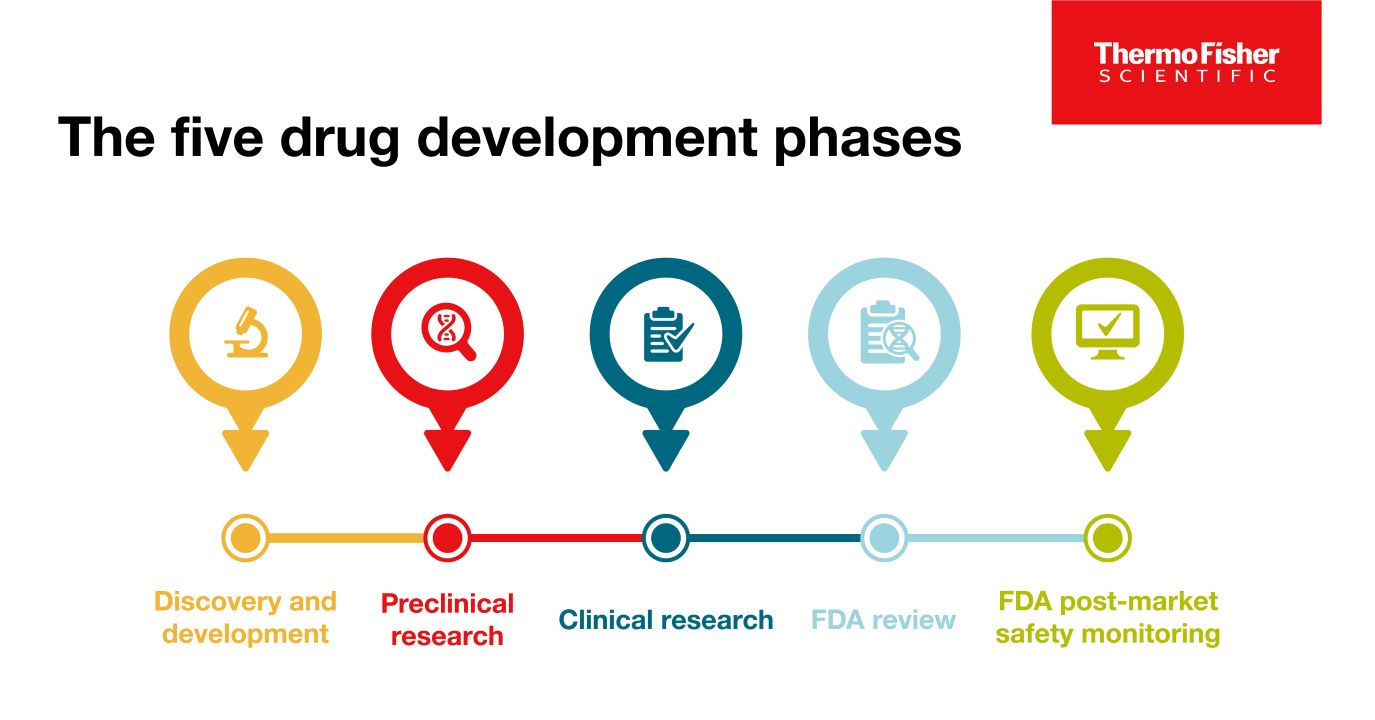
Blog post
The 5 drug development phases
To be deemed a “success,” a new drug must make it through five specific phases: 1) discovery and development, 2) preclinical research, 3) clinical research, 4) FDA review, and 5) safety monitoring.
9 minute read
Blog post
Adherence and Accuracy: Smart packaging advances quality in clinical trials
Learn from Thermo Fisher Scientific’s head of medication adherence and biomarker measurement about data-quality implications of smart packaging and how to integrate them into clinical trials.
15 minute read

Blog post
In Silico Modeling: Accelerating drug development
In silico modeling, both in early development and across the product lifecycle, can streamline drug development and reduce the risks associated with trial-and-error experimental methods. Realizing the potential of the technology requires careful selection and application of in silico strategies and a deep understanding of how to interpret and derive the most valuable insights from the data.
6 minute read
Blog post
Integrated CDMO for Small Molecule Drug Development
A single CDMO partner that offers integrated API, drug product, and clinical strategy activities can streamline and accelerate small molecule drug development. Learn more.
6 minute read

Blog post
Enter the CRDMO: Reshaping drug development through CRO/CDMO integration
CRDMOs, or integrated contract research, development, and manufacturing organizations, are a trend to watch. Discover five benefits of partnering with one.
9 minute read

Blog post
Role of mRNA Encapsulation
mRNA must be encapsulated in different carriers/vectors, such as lipid nanoparticles, to ensure its efficient and effective delivery into target cells. Learn more.
10 minute read
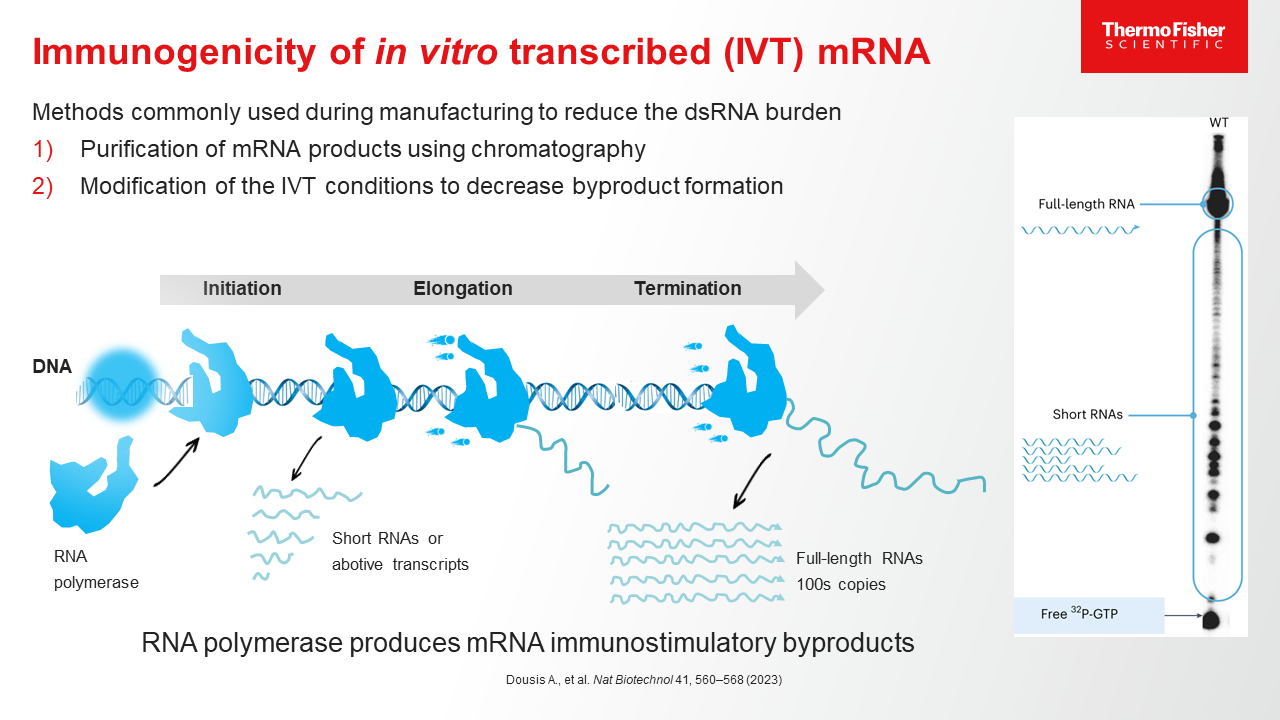
Blog post
mRNA Purification Methods and Process
Learn more about the process of purifying mRNA, and how a CDMO partner with end-to-end mRNA purification experience can help streamline the drug development journey.
10 minute read

Blog post
CROs vs CMOs, and CDMOs: What’s the difference between the three?
CROs, CMOs, and CDMOs all help biotechnology and pharmaceutical companies with drug development and manufacturing, but what’s the difference between the three?
9 minute read
Blog post
Product and partnership quality in viral vector manufacturing: Your gene therapy depends on it
Viral vectors are inherently complex to produce at scale, requiring a laser focus on quality to ensure the efficiency, safety, targeted delivery, and scalability of the gene therapy.
7 minute read

What is a CDMO and what to look for in a partner
Learn how CDMOs (contract development and manufacturing organizations) work with pharma companies, and the top considerations companies have when choosing a CDMO partner.
9 minute read
Blog post
Viral vector commercialization – Part 3: Specialized regulatory support
Find detailed regulatory considerations when preparing viral vectors for commercialization and best practices to address them.
7 minute read
Blog post
Viral vector commercialization – Part 2: Best practices in process validation lifecycle
Learn more about the robust viral vector process validation cycle, which includes various assessments and studies to ensure the safety, efficacy, and quality of viral vectors.
11 minute read
Blog post
Viral vector commercialization – Part 1: Tech transfer process for commercial viral vector manufacturing
Learn how tech transfers can help develop and manufacture viral vectors at scale, accelerate vaccine and gene therapy commercialization, and provide expertise.
9 minute read

Blog post
Top tips for providing the right amount of detail in first-in-human common technical documents
In the early-development stage, little may be known about a drug’s characteristics. What’s more, drug processes and formulations frequently evolve as more information emerges following testing and trials. Learn more.
5 minute read

Blog post
Enabling a digital culture through integrated business processes
In contrast to a physical work environment, where stability and experience are key, a digital business environment focuses on innovation and connectivity. Learn more.
4 minute read
Blog post
Delivering on the promise of cell and gene therapies: A patient-centric approach
In this session, you will gain insights into the complexities of the CGT ecosystem and the challenges that must be overcome to successfully move CGT therapeutics from the laboratory to the patient.
8 minute read

Blog post
Gaining a deeper understanding of your API
As active pharmaceutical ingredients (APIs) become increasingly complex, they pose potential formulation problems that can extend timelines and explode budgets. Read this blog to learn more.
5 minute read

Blog post
EU and US regulations: What’s coming for cell and gene therapies?
Cell and gene therapy (CGT) developers today face an added challenge in their quest to bring a product through clinical trials and to the market. Read this blog to learn more.
7 minute read
Blog post
Choosing a CDMO for mRNA success: Five CDMO characteristics needed
The promise of mRNA technologies has been clearly demonstrated during the COVID-19 pandemic, with vaccines reaching the market in record time. The vital role...
7 minute read
Blog post
Trends in mRNA therapeutics: Pandemic learnings for a pathway to success
The rapid advancement of the Pfizer-BioNTech and Moderna messenger RNA–based COVID-19 vaccines from lab to clinic—with development taking less than one year—has validated the...
5 minute read
Blog post
Moving from vials to prefilled syringes for vaccines: Three key success factors
As pharmaceutical companies become more patient-centric and self-administration of injectable drugs continues to increase, the market for drug products in prefilled syringes is forecast to grow, reaching $9.53 billion by 2026.
4 minute read

Blog post
The Four Stages of Equipment Qualification
As discussed in my previous blog, qualification is the process of establishing documented evidence that a specific equipment, facility or system are fit and ready for their intended use.
3 minute read

Blog post
Myths & Facts about Ancillaries
It’s a fact that ancillary supplies are frequently perceived as less important than study drug.
2 minute read

Blog post
Is Your Supply Chain Bulletproof?
Before you respond, think hurricanes, Nor’easters and tsunamis. Earthquakes, typhoons and volcanic eruptions. Civil unrest and war. Terrorism. A pandemic virus.
2 minute read

Blog post
Top Tips on How to Manage Clinical Label Translation and Regulatory Requirements
Wouldn’t it be nice to find a way to stay on top of label translation and regulatory requirements for every country included in your clinical trial?
2 minute read

Blog post
Introducing an Expanded Packaging Service for Specialty Products
Developing a specialty drug for a complex or rare disease is an achievement worthy of celebration.
2 minute read
Blog post
Is it Time to Start Thinking about Packaging?
If this headline caught your eye, it may be because you’ve received promising pre-clinical results (Congratulations!) and you’re starting to think about planning your next steps.
2 minute read
Blog post
Taking Your API to the Next Level: Three Steps to Consider Before Outsourcing
With outsourcing API development becoming more common, we see a rise of multiple competing Contract Development Manufacturing Organizations (CDMOs) as potential development partners.
10 minute read

Blog post
Break Through the OTC Noise
It’s no secret—consumers have a vast range of OTC options at their favorite in-store or online retailer. Everything from tablets and capsules, to syrups—consumers have more options than ever in the OTC jungle.
8 minute read
Blog post
Considerations and roadblocks that stifle orphan drug development
According to the US Food and Drug Administration (FDA), “2020 was a record-breaking year in terms of the number of orphan drug designation and rare pediatric disease designation requests submitted to the Office of Orphan Products Development.”
6 minute read
Blog post
Regulatory landscape in Europe: Key advice for meeting post-Brexit Qualified Person requirements
The United Kingdom’s (UK) departure from the European Union (EU) has added a layer of complexity to the clinical trial supply chain in Europe above and beyond COVID-related disruptions.
(4 minute read)

Blog post
Time to embrace electronic labels? Potential labeling solutions under the new EU Clinical Trial Regulation
The new EU Clinical Trial Regulation (CTR) is intended to simplify clinical trial administration and create a more welcoming climate for pharmaceutical companies that operate in Europe.
5 minute read

Blog post
Navigating the Complexities of Process Performance Qualification
Method qualification is monumentally important before process performance qualification (PPQ). This early assessment of your method’s performance characteristics is critical as it pertains to method validation and its parameters such as precision, accuracy, and linearity.
5 minute read

Blog post
Continuous Manufacturing: An Efficient Way to Produce OSD Drugs
When Henry Ford revolutionized manufacturing practices back in 1913 in Highland Park, MI, his main goal was simple—to make the best possible product in the most efficient and cost-effective manner. Ford’s focus on bettering the “flow” of manufacturing to enable workers/technology to work smarter and reduce waste of raw materials, changed manufacturing principles forever.
6 minute read
Blog post
COVID-19’s Silver Lining: Accelerated Vaccine Development
Vaccine development is a lengthy process—it is expensive, attrition is high, and to get a licensed vaccine to everyone, it takes multiple candidate iterations. Vaccine development for pandemics and epidemics is risky, and due to the novel nature of viruses, certain unknown factors can derail a vaccine program.
6 minute read
Blog post
A Day in the Life of a Viral Vector Partner
When it comes to a viral vector Contract and Development Manufacturing Organization (CDMO), what sort of qualities should they possess?
9 minute read

Blog post
How Decentralized Clinical Trials Enhance Patient-Centricity in the Age of COVID-19
As COVID-19 continues to change how we do business in the biopharmaceutical industry, it’s important to not lose sight of why we do what we do: improving and saving patient lives.
5 minute read

Blog post
Work Smarter, Not Harder: Accelerating Your Biologics Development and Commercialization
Over the past decade, the biologics industry has seen double digit growth and an overall increase of market share.
5 minute read

Blog post
Prefilled Syringes: Three Pain Points You May Not Have Considered
As pharmaceutical companies look to become more patient-centric, certain drug products come to the forefront to support that effort.
5 minute read
Blog post
Navigating Cell & Gene Therapy Regulations: How Does Your CDMO Match Up?
Whether you are a large or new and emerging biotech company, many companies find themselves lacking the internal resources and/or expertise to properly support regulatory submissions.
7 minute read

Blog post
Choosing a CDMO Who is a True Bioproduction Expert
With the number of CDMO’s rising in the biologics manufacturing industry, it can be challenging for new and emerging biopharmaceutical companies to determine which CDMO is right for them.
8 minute read